Protocol p. 20
An estimated 15 to 24 patients will be enrolled in Phase 1b, depending on dose escalation and expansion.
Protocol p. 27
An estimated 32 patients will be enrolled in Phase 1b, depending on dose escalation and expansion.
Using our AI pipeline, we've analyzed thousands of publicly available clinical trial protocols. We've found inconsistent language, ambiguities, and knowledge gaps. All of which compromise the ability to run a trial.
Unlike other approaches that treat the protocol as a simple input-output problem, we believe the complexity and stakes of clinical trials demand a quality copilot that can help human teams understand and improve their protocols.
Tufts CSDD conducted a recent study and cited this quote from a survey participant:
We often are receiving protocols, lab manuals, IB, pharmacy manuals, ICF—that are not in agreement with each other or have glaring mistakes that are not corrected. Study procedure timelines don't match the lab processing for those study procedures, or data collection guides don't match the visit schedule. We often see inclusion and exclusion criteria that contradict each other. All of this adds confusion, unavoidable PDs (protocol deviations), missing data, out of window study visits, but most importantly it can impact patient safety and data quality.
Source: Harper, B., Ford, R. M., Krayem, R., Nomizu, R., Andrus, J., & Getz, K.
Characterizing the Protocol-Guided Source Preparation Process at Investigative Sites.
AI accelerates content creation and systems configurations, so that studies can be built in a fraction of time. But an inconsistency that once slowed a single site now spreads to every connected system at machine speed - unreviewed, everywhere at once.
of Phase I-IV protocols now require at least one substantial amendment
of those amendments are considered avoidable
direct cost of a single substantial amendment, by complexity
average added timeline per amendment
On average, a phase 3 trial carries 5-7 substantial amendments with each one affecting dozens of downstream artifacts, decisions and regulated systems. HumanTrue is the verification layer that is with you end-to-end, propagating the needed changes with built in re-verification before final release.
HumanTrue provides the details that you need to operationalize an amendment.
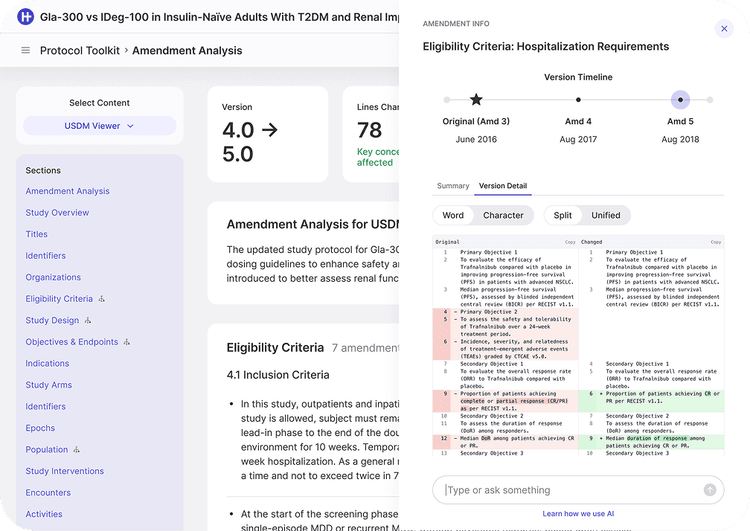
A paragraph-by-paragraph delta of the protocol itself, with attribution to author and version.
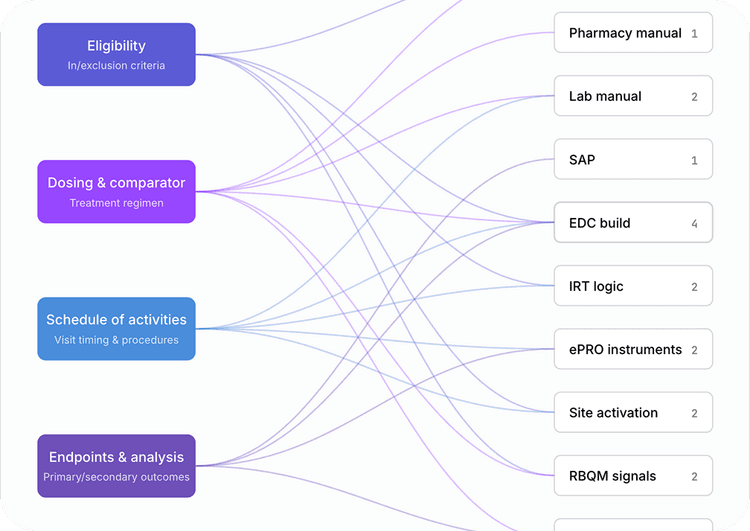
Every downstream artifact, system, and site instruction the amendment touches visualized as a propagation tree.
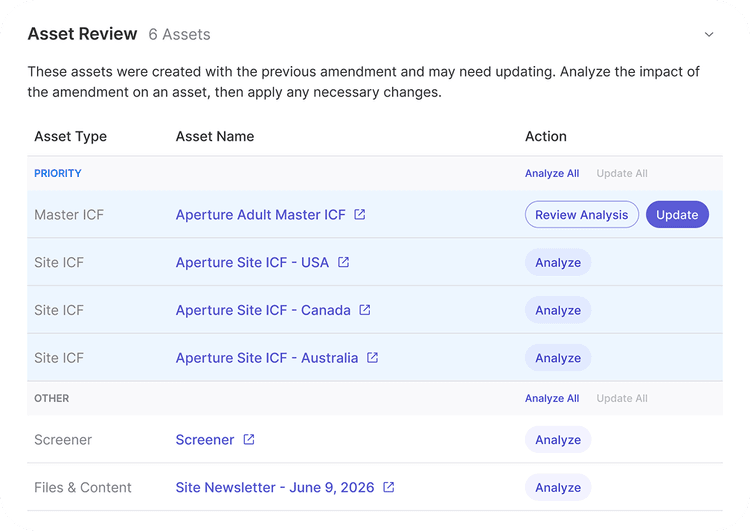
Affected sections of every downstream document regenerated against the new source with citations.
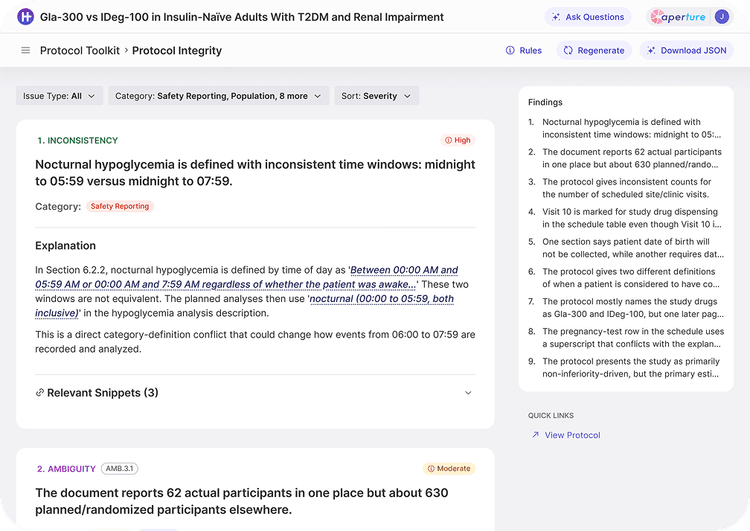
Every artifact in the chain is re-checked against the new source, protecting against silent drift between the protocol and downstream documents.
HumanTrue gives a lean team the roster of a global sponsor. Every protocol, site, and amendment runs through the same verified semantic model - so a handful of people get the rigor that used to take a department of specialists, on every protocol, ICF, IRB submission, and amendment alike. The expertise doesn't have to scale with your headcount. It's already built in.
Per-protocol QA at scale. AI-assisted verification built-in; human certified review available.
Operational services. No internal build needed. You get protocol verification, amendment governance, and startup-package generation from day one.
From submission to site. Everything from regulatory filings to site-executable materials is produced from one verified semantic model, with full provenance on every output.
Speed-to-activation. Coherent protocols mean faster activation and far less avoidable cost.
Partner-grade trust. CRO integrations, IRB references, and source in every output. The deliverable a sponsor partner expects.
